Application de l’Analyse en Composantes Principales pour étudier l’adaptation biologique en génomique des populations.
Keurcien LUU
21 décembre 2017
Chapitre 1 Introduction
1.1 La génétique des populations
“Il est bon de rappeler que ce qui nous rend semblables est plus important que ce qui nous rend différents. Les milliards d’êtres humains éparpillés sur toute la planète se différencient par la couleur de la peau et par la forme du corps, par la langue et par la culture. Et cette variété, qui témoigne de notre capacité à changer, à nous adapter à des milieux différents et à y développer des modes de vie originaux, est la meilleure garantie pour l’avenir de l’espèce humaine. Les connaissances que nous avons acquises sur nous-mêmes montrent cependant que toute cette diversité, comme la surface changeante des océans ou de la voûte du ciel, est bien peu de chose par rapport à cet immense patrimoine que nous avons en commun, nous, les humains.”
— L. Cavalli-Sforza (1994)
1.1.1 L’évolution comme point de départ
« La génétique est la science de l’hérédité. Elle est la clé de toute la biologie, parce qu’elle explique les mécanismes qui sont responsables de la reproduction des êtres vivants, du fonctionnement et de la transmission du matériel héréditaire, des différences entre les individus, de l’évolution biologique. »
Cette définition, donnée par Cavalli-Sforza et traduite ici de l’italien par Françoise Brun (L. Cavalli-Sforza, 1994), restitue également les motivations à l’origine de l’émergence du domaine de la génétique des populations, à savoir l’étude de la variabilité interindividuelle d’un point de vue évolutionniste. Pour John H. Gillespie, il s’agit de la « discipline qui fait le lien entre la génétique et l’évolution » (Gillespie, 2010) : « La génétique des populations s’intéresse à l’évolution d’un point de vue génétique. Elle diffère de la biologie en ce que ses idées les plus importantes ne sont pas expérimentales ou observationnelles mais davantage théoriques. Il pourrait difficilement en être autrement. Les objets d’étude sont principalement la fréquence et la valeur sélective des génotypes dans les populations naturelles. »
Malgré cette caractérisation, les fondements de la génétique des populations trouvent en réalité leurs origines bien avant la formalisation en 1909 par Wilhelm Johannsen du concept même de gène (Roll-Hansen, 2014), en témoignent les travaux de Charles Darwin (1809-1882) et de Gregor Mendel (1822-1884). L’Origine des espèces, publié en 1859 et considéré encore à ce jour comme le texte fondateur de la théorie de l’évolution (Darwin, 1980), énonce les premiers principes de la sélection naturelle. Les travaux de Mendel, figurent quant à eux parmi les premiers à se pencher sur les mécanismes de l’hérédité d’un point de vue statistique, notamment via l’étude de phénotypes en termes de proportions et de fréquences.
1.2 À l’origine de la variabilité génétique
1.2.1 La théorie de l’évolution
En 1859, Darwin soutenait l’idée selon laquelle la principale force évolutive est la sélection naturelle (Darwin, 1980). « Je me propose de passer brièvement en revue les progrès de l’opinion relativement à l’origine des espèces. Jusque tout récemment, la plupart des naturalistes croyaient que les espèces sont des productions immuables créées séparément. De nombreux savants ont habilement soutenu cette hypothèse. Quelques autres, au contraire, ont admis que les espèces éprouvent des modifications et que les formes actuelles descendent de formes préexistantes par voie de génération régulière. »
C’est de cette manière qu’en 1920, Edmond Barbier, dans sa notice relative à la traduction française de L’Origine des espèces (Darwin, 1980), décide de présenter le contexte dans lequel il a été amené à effectuer ce travail de traduction. Bien que la théorie de Darwin fut globalement bien accueillie par la communauté scientifique, elle fut tout de même en proie à de nombreuses critiques. L’une des principales critiques émises à son encontre fut relative à la croyance de Darwin selon laquelle l’hérédité par mélange serait le principal mode de transmission des caractères héréditaires (Gayon, 1992). Or, si sélection naturelle il y a, la conservation et la transmission des caractères sélectionnés sont essentielles. Si bien qu’une hérédité par mélange n’est pas envisageable pour soutenir la thèse de la sélection naturelle, puisque tout caractère transmis de cette façon se verrait altéré (ou dilué si l’on souhaite conserver l’idée de mélange) à chaque génération et donc éliminé après un certain temps. Cependant, sa théorie bénéficiera par la suite des travaux de Mendel qui, lors de leur redécouverte en 1902 (Bateson & Mendel, 1913), apporteront l’élément fondamental manquant à la théorie darwinienne : le principe d’hérédité mendélienne. Cette théorie de l’évolution néo-darwinienne, née de la conciliation de la théorie darwinienne et du principe d’hérédité de Mendel, constitue le paradigme évolutionniste tel que nous le connaissons aujourd’hui et porte le nom de théorie synthétique de l’évolution.
1.2.2 L’évolution d’une théorie
À la théorie néo-darwinienne est souvent opposée la théorie neutraliste développée par Motoo Kimura dans son ouvrage The neutral theory of molecular evolution (Kimura, 1983), bien que ces deux théories ne soient pas incompatibles. La première suggère que les mutations apparaissent à la faveur de la sélection naturelle. La seconde affirme quant à elle que l’évolution ne serait que le résultat de mutations qui surviennent de façon tout à fait aléatoire, tout en étant sélectionnées selon le même mécanisme de sélection naturelle proposé par Darwin (Figure 1.1).
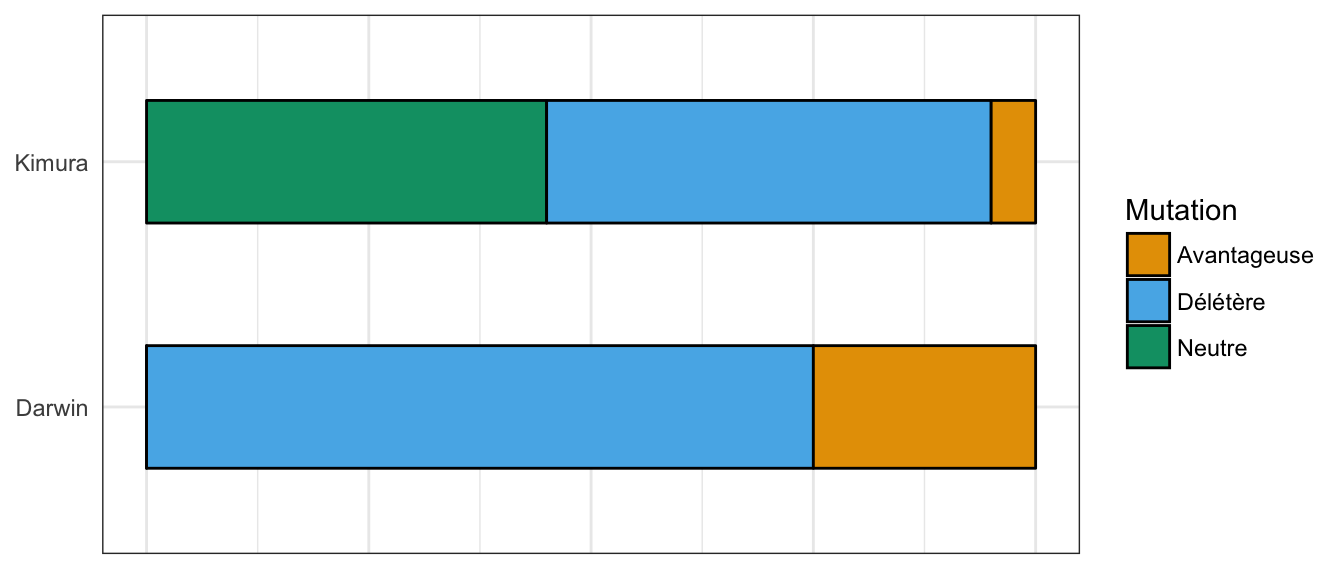
Figure 1.1: Représentation schématique des probabilités d’occurrence pour chaque type de mutation pour la théorie sélectionniste de Darwin et pour la théorie neutraliste de Kimura (Bromham & Penny, 2003). Selon la théorie de Darwin, la plupart des mutations sont délétères et le reste des mutations confère un avantage sélectif. Selon la théorie de Kimura, une partie des mutations qui apparaissent n’ont pas d’effet sur la valeur sélective. Ces mutations sont dites neutres.
Une des composantes principales de cette nouvelle théorie consiste à affirmer que les fluctuations aléatoires dans les fréquences d’allèle, n’affectant que très peu ou pas du tout la valeur sélective, constituent la principale source de variabilité de l’ADN (B. Charlesworth & Charlesworth, 2009). Une grande partie de la variation génétique observée est fonctionnellement neutre et n’occasionne pas de changement de valeur sélective.
L’approbation de cette théorie, bien que conceptuellement intéressante, aura un retentissement beaucoup plus important d’un point de vue de la méthodologie statistique. La formulation d’une hypothèse permettant de décrire un processus évolutif en l’absence de sélection, portant généralement le nom d’hypothèse nulle ou encore de modèle neutre, est souvent de première nécessité dans toute démarche visant à caractériser un mécanisme de sélection. La donnée d’observations mettant en défaut le modèle neutre aura pour conséquences de créditer davantage une hypothèse invoquant un processus de sélection. Historiquement, la statistique \(D\) de Tajima fut l’une des premières statistiques développées à partir d’une hypothèse nulle bâtie pour les mutations neutres (Tajima, 1989).
1.2.3 Forces évolutives
Ce changement de paradigme nous invite de ce fait à observer la sélection naturelle à travers le prisme de la théorie neutraliste, et donc à identifier les mutations sélectives comme des mutations dont les origines ne peuvent être uniquement expliquées par des processus biologiquement neutres. La génétique des populations distingue trois types de processus neutres : la dérive génétique, les mutations aléatoires et le flux de gènes. Ces processus, tout comme la sélection naturelle, constituent les principales forces évolutives.
“La mutation propose, la sélection dispose.”
— L. Cavalli-Sforza (1994)
La dérive génétique
La dérive génétique correspond à tout ce qu’il y a d’aléatoire dans l’évolution d’une population. C’est le hasard des choses. Le nombre de descendants ou le choix de partenaire sexuel sont des exemples de phénomènes aléatoires participant à la dérive génétique. Le principe de dérive génétique est illustré en figure 1.2, à l’aide du modèle de Wright-Fisher tel qu’il est présenté dans l’ouvrage Population Genetics (Gillespie, 2010). Ce modèle simpliste suppose que la taille de la population est constante et que chaque individu de la \({n+1}^\text{ème}\) génération est issu du brassage génétique de deux individus tirés aléatoirement de la \(n^\text{ème}\) génération.
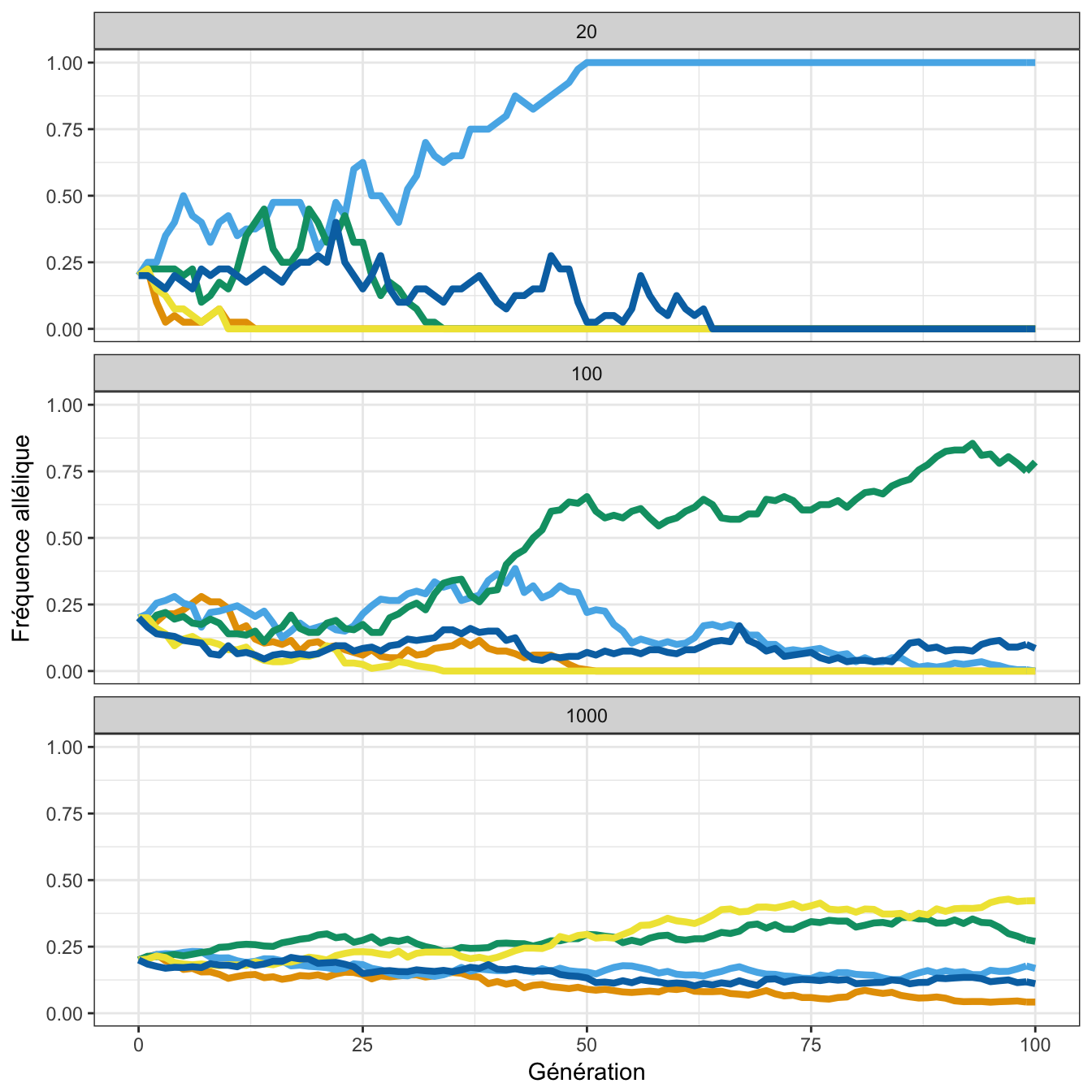
Figure 1.2: Simulation numérique de la dérive génétique à l’aide du modèle de Wright-Fisher. La fréquence de l’allèle étudié est simulée pour 5 populations constituées chacune de 20, 100 ou 1000 individus sur une période de 100 générations. Dans chaque population, la fréquence de l’allèle est initialement de 0.20 (Gillespie, 2010).
En particulier, la figure 1.2 met en évidence deux caractéristiques de la dérive génétique :
Les fréquences alléliques évoluent de façon indépendante d’une population à une autre.
Pour un nombre de générations fixé, la dérive génétique entraîne une perte de diversité allélique plus rapidement au sein des populations de plus petite taille. Dans le modèle de Wright-Fisher, les fréquences alléliques finissent éventuellement par atteindre les états dits absorbants que sont 0 et 1.
Les mutations aléatoires
Si la dérive génétique entraîne une perte de diversité allélique, les mutations favorisent quant à elles le maintien des variations génétiques entre les populations (Gillespie, 2010). Les mutations apparaissent principalement lors de la phase de réplication de l’ADN. Une mutation peut survenir à un locus donné avec une probabilité spécifique à chaque espèce (J. W. Drake, Charlesworth, Charlesworth, & Crow, 1998), appelée taux de mutation (Table 1.1).
| Espèce | Taux de mutation |
|---|---|
| \(E.\) \(coli\) | 5.4e-10 |
| \(C.\) \(elegans\) | 2.3e-10 |
| Drosophile | 3.4e-10 |
| Souris | 1.8e-10 |
| Homme | 5.0e-11 |
Le flux de gènes
Le flux de gènes est généralement le résultat d’évènements migratoires initiés par des individus appartenant à une population donnée, vers une seconde population dont les fréquences d’allèles diffèrent éventuellement de la population d’origine. Le flux de gènes influe sur la diversité génétique initialement présente si par exemple, parmi les allèles migrants figurent des allèles qui n’existaient pas dans la population receveuse.
La sélection naturelle
En biologie évolutive, la sélection naturelle est la force qui tend à préserver les allèles conférant des avantages quant à la viabilité ou la fertilité d’un individu. Elle agit sur les traits qui sont héritables. La grande majorité de ces traits sont transmis aux descendants en suivant le mécanisme d’hérédité génétique.
1.2.4 Adaptation locale
1.2.4.1 Principe
La diversité climatique et la diversité géologique terrestre ont naturellement façonné des environnements aux constitutions physiques et chimiques variées. À celles-ci viennent s’ajouter des caractéristiques écologiques résultant des interactions entre l’environnement et les organismes qui y évoluent lato sensu. La valeur sélective de ces organismes, désignant leur capacité de survie et de reproduction, peut être impactée par les caractéristiques environnementales auxquelles ils sont exposés. Si cette valeur sélective est associée à un trait phénotypique, on dit que ce trait confère un avantage adaptatif et on parle dans ce cas d’adaptation locale. Une population sera ainsi dite adaptée à son environnement si elle développe, par le biais de la sélection naturelle, un ou plusieurs allèles associés à un trait adaptatif augmentant la valeur sélective des individus la constituant. Ceci en réponse aux pressions environnementales auxquelles ces individus sont soumis. À titre d’exemple, nous pouvons citer l’adaptation des populations tibétaines et andines à la haute altitude (Table 1.2).
| Trait | Andins | Tibétains |
|---|---|---|
| Augmentation du taux d’hémoglobine | oui | à partir de 4000m d’altitude |
| Augmentation de la pression artérielle pulmonaire | oui | non |
| Augmentation de la ventilation au repos | oui | non |
| Prévalence du mal chronique des montagnes | 5% | 1% |
Chez l’Homme moderne, les habitudes alimentaires et les modes de vie constituent également des caractéristiques environnementales importantes. L’agriculture et le pastoralisme1 ont notamment participé à la diversification des environnements humains (Jeong & Di Rienzo, 2014). En Europe, l’adaptation biologique à ces nouveaux modes de vie s’est par ailleurs manifesté par la sélection du phénotype LP, dit de persistence de la lactase, caractérisant l’aptitude à digérer le lactose à l’âge adulte (Itan, Powell, Beaumont, Burger, & Thomas, 2009).
1.2.4.2 L’hybridation et le flux de gènes comme sources d’adaptation
Un caractère (un variant allélique par exemple) peut être sélectionné au sein d’une population de différentes manières :
le caractère y est déjà présent et devient adaptif suite à un changement d’environnement (standing genetic variation).
le caractère y apparaît à la suite d’une mutation spontanée (de novo mutations).
le caractère est hérité d’une autre population par flux de gènes (adaptive introgression).
Cette dernière possibilité est d’ailleurs très intéressante pour une espèce d’un point de vue de la diversité génétique. Pour enrichir le catalogue d’allèles, nous avons vu plus haut que le flux de gènes constituait un processus clé. La reproduction sexuée est connue pour jouer un rôle important dans la sélection naturelle, offrant aux individus la possibilité, par le biais du brassage allélique, d’hériter des “meilleurs allèles” que possèdent leurs parents. Augmenter la diversité génétique augmente ainsi les chances de voir se transmettre des allèles favorables.
Nous parlons d’hybridation ou de métissage lorsque deux individus, appartenant à deux populations différentes, se reproduisent entre eux. Pour ce qui est de la distinction de populations, nous nous en tiendrons à la suggestion faite par Harrison & others (1990), considérant que deux individus issus de populations différentes doivent chacun posséder des traits héritables qui les différencient. Ainsi, l’hybridation constitue un excellent moyen pour permettre à une espèce d’intégrer de nouveaux allèles (Stevison, 2008). Bien que l’hybridation conduise fréquemment à la naissance d’individus à la valeur sélective hautement diminuée voire d’individus non fertiles, il arrive que certains descendants aient tout de même la capacité de se reproduire et ainsi être à l’origine de l’apparition de nouveaux allèles. La sélection de tels allèles transmis par flux de gènes ou par hybridation correspond à ce que l’on appelle l’introgression adaptative. Des exemples d’introgression sont observés à la fois dans la nature (introgression naturelle) et à la fois chez des espèces domestiquées (introgression délibérée). Nous pouvons citer l’exemple de la souris domestique, Mus musculus domesticus, qui a hérité d’un gène de résistance à la coumadine (communément appelée « mort aux rats ») en s’hybridant avec une espèce sauvage, Mus spretus (Y. Song et al., 2011). Ce gène de résistance est localisé sur le chromosome 7 de la souris domestique et appartient à une région qui a été identifiée comme provenant effectivement de la souris sauvage.
L’identification des mutations génétiques responsables de l’adaptation est particulièrement cruciale pour la compréhension des processus évolutifs et plus spécifiquement ceux liés à la spéciation2. L’introgression adaptative permet quant à elle de comprendre l’adaptation rapide de certaines espèces et en quoi l’hybridation constitue un vecteur important pour l’adaptation (Hufford et al., 2013). Bien que l’étude de l’adaptation locale puisse être menée de façon expérimentale par le biais de la transplantation réciproque (Kawecki & Ebert, 2004), nous proposons ici d’exploiter les génotypes de populations naturelles.
1.3 Données de polymorphismes génétiques
Avant d’aborder la partie sur les méthodes développées au cours de cette thèse, nous donnons une rapide présentation du type de données que nous traitons, à savoir des données de séquençage. Le séquençage de l’ADN consiste à déterminer l’ordre dans lequel sont agencées les paires de bases pour un fragment d’ADN donné. Son apparition a offert de nouvelles voies d’exploration en biologie évolutive. Les variations génétiques étaient jusqu’alors appréciées par le biais des différences phénotypiques. En phylogénie par exemple, le séquençage de l’ADN a donné naissance à la phylogénétique moléculaire, qui se distingue de la phylogénétique traditionnelle en ce qu’elle ne considère que les séquences de nucléotides pour évaluer la proximité entre deux espèces. Et c’est précisément l’accès à ces séquences qui va permettre à toute une population de méthodologies statistiques de voir le jour et de se développer.
1.3.1 Des données en grande dimension
Le séquençage nouvelle génération (appelé encore séquençage à haut débit ou NGS) a connu un essor considérable au cours des dernières décennies. Si bien que les prouesses techniques et les progrès technologiques réalisés dans ce domaine ont permis de réduire d’un facteur 100,000 les coûts de séquençage en l’espace de seulement 15 ans (Figure 1.3).
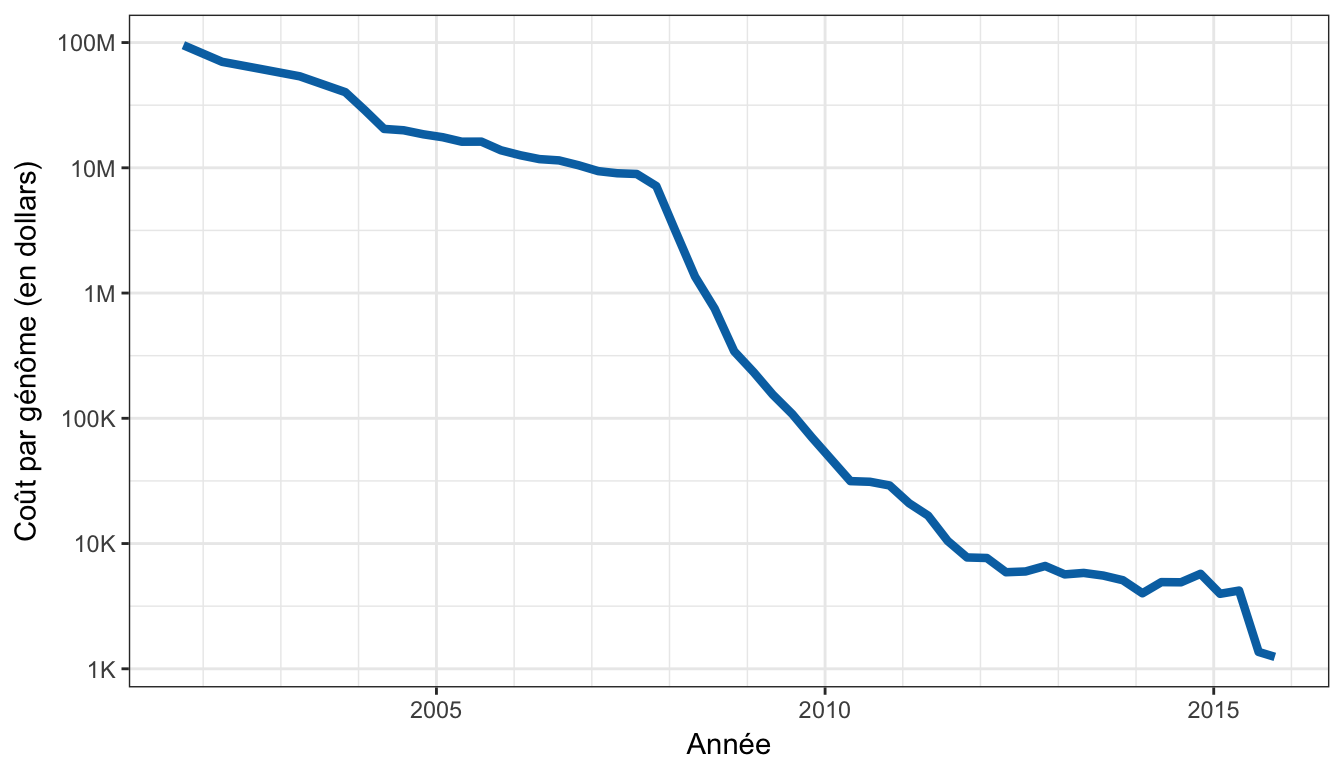
Figure 1.3: Évolution des coûts de séquençage depuis 2001 (Wetterstrand, 2013).
Toutefois, compte tenu de la popularité croissante des technologies NGS (Muir et al., 2016) et des considérables volumes de données qu’elles génèrent, de nouvelles problématiques se posent quant à leur stockage et leur analyse, nécessitant l’utilisation de puissantes ressources de calcul ainsi que le développement d’algorithmes plus adaptés (Gogol-Döring & Chen, 2012).
1.3.2 Les marqueurs génétiques
Le séquençage de l’ADN a également permis de faire évoluer le concept de marqueur génétique. Un marqueur génétique correspondait autrefois à un gène polymorphe3 identifié sur la base d’observations phénotypiques. Grâce au séquençage de l’ADN, une nouvelle définition tenant compte de la position sur le chromosome a été adoptée pour caractériser un marqueur génétique. Différents types de marqueurs génétiques ont été identifiés, parmi lesquels figurent les microsatellites, les insertions, les délétions et les SNPs4. La structure spatiale de l’ADN n’étant pas prise en compte dans les travaux présentés ici, nous en garderons une représentation unidimensionnelle.
Microsatellite
Jusqu’à présent, les microsatellites ont connu un succès important, notamment grâce à la popularisation de techniques telles que la PCR (Réaction en Chaîne par Polymérase). Cependant, grâce aux nouvelles avancées technologiques que nous évoquerons un peu plus loin, ils sont progressivement délaissés au profit des SNPs. Un microsatellite est repérable par la répétition successive de petits motifs chacun composé de 1 à 4 nucléotides (Figure 1.4).

Figure 1.4: Exemples de microsatellites. La première séquence comporte 3 répétitions du motif CCG, tandis que la seconde inclut 4 répétitions du motif CA.
Insertion/Délétion (Indel)
L’insertion ou la délétion d’une base constituent également des polymorphismes génétiques. Relativement à une séquence de nucléotides de référence, une insertion consiste en la présence d’une base supplémentaire tandis que la délétion consiste en l’absence d’une base (Figure 1.5).
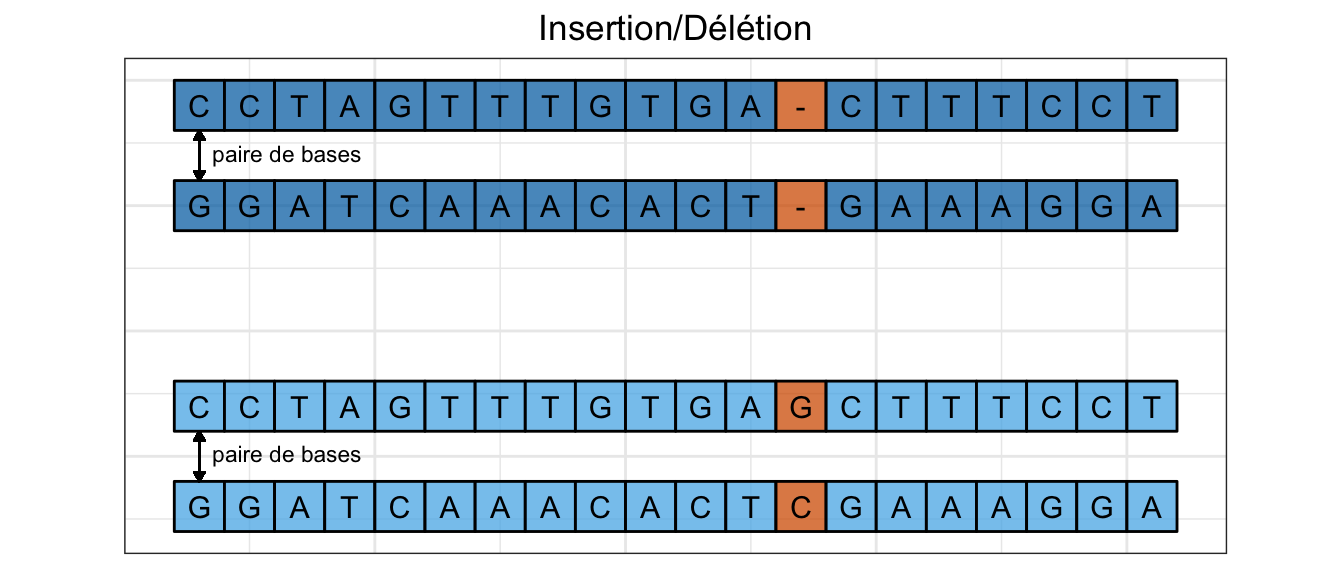
Figure 1.5: Exemple d’Indel.
Polymorphisme d’un seul nucléotide (SNP)
Le polymorphisme d’un seul nucléotide correspond au polymorphisme génétique le plus simple, et correspond à l’emplacement d’un nucléotide présentant des variations appréciables à l’échelle d’une population (Figure 1.6).
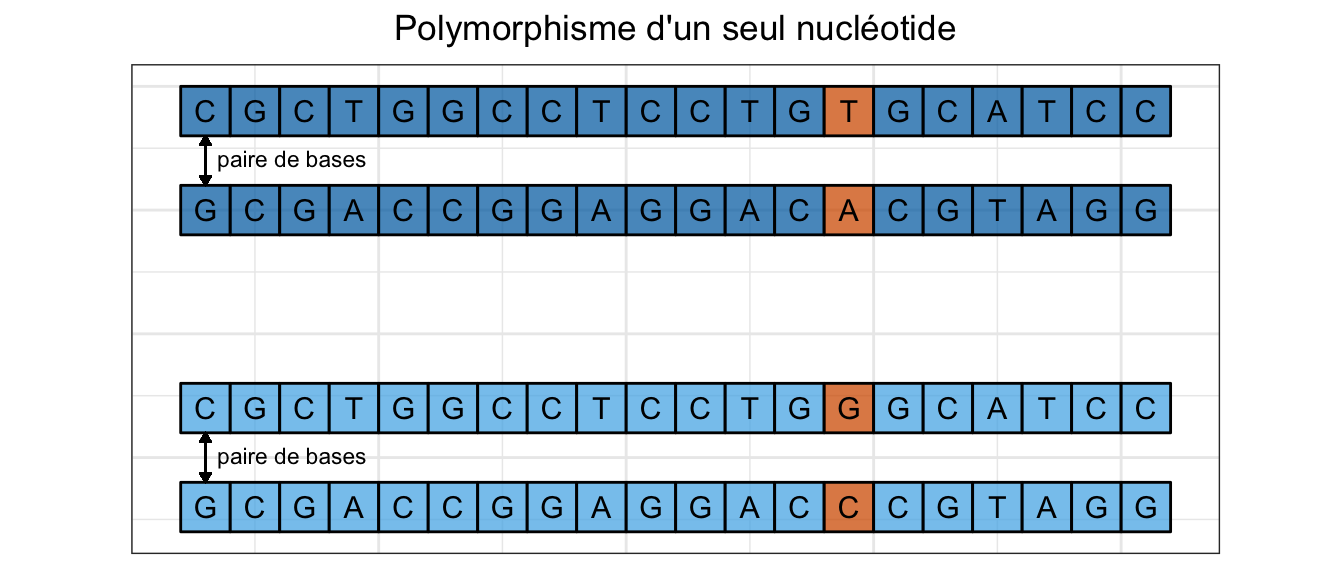
Figure 1.6: Exemple de SNP.
1.3.3 Encodage des données génétiques
Dans ce paragraphe nous présentons le format des données sur lesquelles nous travaillons et définissons quelques notations qui seront utilisées par la suite. Conformément à l’usage qui en est fait par Gillespie (2010), nous emploierons le terme d’allèle pour désigner la version d’une base nucléique à un locus donné. Dans le cadre de nos travaux, nous considèrerons que pour un locus donné, il n’existe que deux nucléotides possibles (SNP bi-allélique), dont l’un sera considéré comme l’allèle de référence et l’autre comme l’allèle alternatif. Pour un locus donné, l’encodage des données de SNPs consiste à attribuer à chaque individu (Figure 1.7) :
la valeur \(0\) s’il est homozygote pour l’allèle de référence.
la valeur \(1\) s’il est hétérozygote.
la valeur \(2\) s’il est homozygote pour l’allèle alternatif.
Une base de données de génômes constituée de séquences de nucléotides pourra donc en pratique être encodée par une matrice, appelée matrice de génotypes, composée uniquement de \(0\), \(1\) et \(2\).
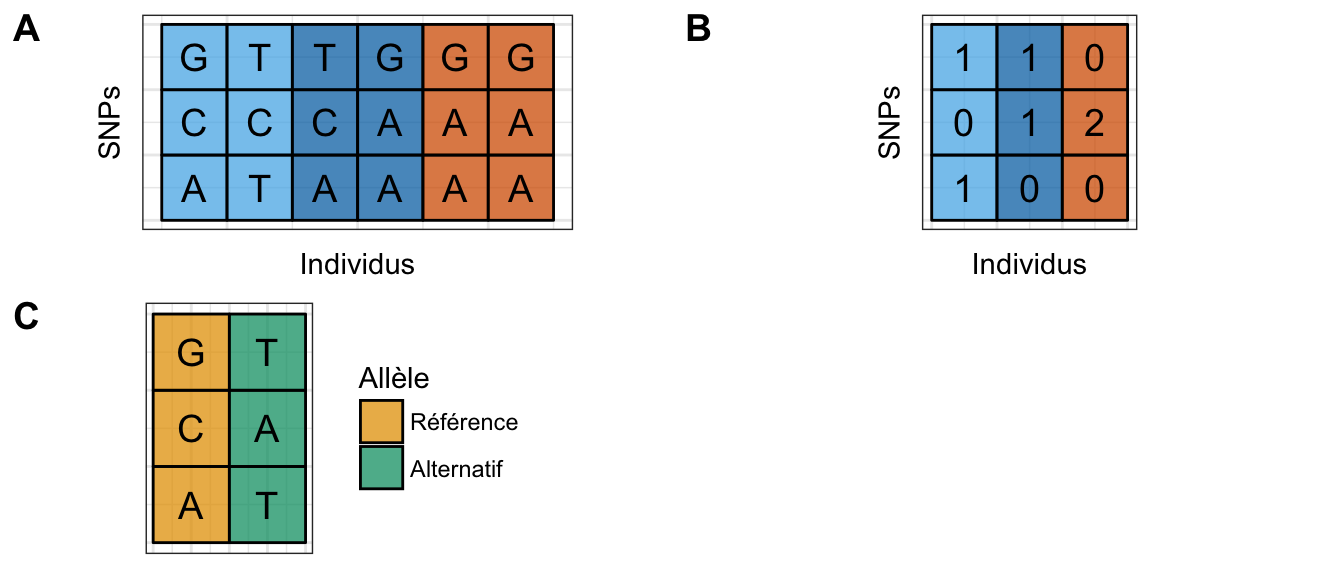
Figure 1.7: Exemple d’encodage de données de SNPs. A. Chaque ligne de la matrice correspond à un SNP et chaque individu est représenté par une paire de colonnes (de la même couleur). B. Matrice de génotypes résultant de l’encodage. C. Chaque ligne de la matrice correspond à un SNP. La première colonne indique quel allèle est considéré comme allèle de référence. La seconde colonne indique quel allèle est considéré comme allèle alternatif.
Dans la mesure du possible, l’allèle de référence (resp. alternatif) est choisi de façon à correspondre à l’allèle ancestral (resp. dérivé). En pratique, le choix de l’allèle de référence et de l’allèle alternatif peut se faire de façon totalement arbitraire sans que cela n’influe sur les méthodes statistiques basées sur la variance des allèles, ce qui sera généralement le cas pour celles qui sont présentées ici.
Les matrices de génotypes seront généralement notées \(G\) et seront considérées comme des éléments de \(\mathcal{M}_{np}(\{0,1,2\})\), où \(p\) désigne le nombre de locus et \(n\) le nombre d’individus. Ainsi, \(G_{i,.}\) désignera le vecteur de taille \(p\) composé du génotype de l’individu \(i\). De même, \(G_{.,j}\) désignera le vecteur de taille \(n\) contenant les comptages de l’allèle alternatif pour les différents individus au locus \(j\). À la matrice \(G\) sera associée la matrice \(\tilde{G}\), normalisée pour les lignes (c’est-à-dire pour les SNPs). Notant \(f_j\) la fréquence allélique de l’allèle alternatif (ou de l’allèle de référence), nous définissons \(\tilde{G}_{.,j}\) par :
\[\begin{equation} \tilde{G}_{.,j} = \frac{G_{.,j} - 2f_j}{\sqrt{2f_j(1-f_j)}} \end{equation}\]1.4 Les motivations de la thèse
La production de données génétiques, volumineuses par la quantité d’information qu’elles renferment, laisse présager le meilleur pour les domaines de la médecine clinique et de la biologie évolutive. En génétique des populations, leur acquisition offre de nombreuses perspectives d’étude, notamment concernant la mise en évidence de gènes impliqués dans les processus évolutifs ou de gènes associés à certains phénotypes. Les méthodes répondant à cette problématique portent le nom de scans génomiques. Les scans génomiques pour la sélection, destinés à isoler des locus sous sélection, se sont largement répandus au cours des dernières années, principalement grâce au développement fulgurant qu’ont connu les technologies NGS. Pour illustrer le principe des scans génomiques, nous reprenons l’exemple de l’adaptation à l’altitude des populations tibétaines.
Pour détecter les gènes responsables de cette adaptation, Xu et al. (2010) ont réalisé un scan génomique en analysant la différenciation génétique entre une population constituée de tibétains et une autre constituée de Hans. Le résultat de ce scan génomique5 est présenté en figure 1.8.
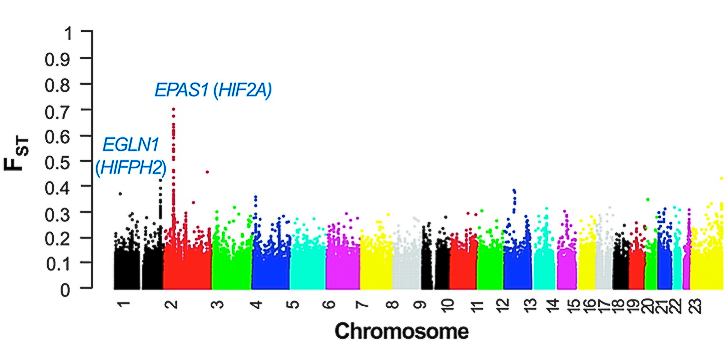
Figure 1.8: Scan génomique réalisé sur des populations de tibétains et de Hans utilisant la \(F_{ST}\) (mesure de différenciation génétique de référence que nous introduirons dans le chapitre suivant). Deux régions génomiques présentant des valeurs de \(F_{ST}\) significativement élevées ont été identifiées sur les chromosomes 1 et 2 (Xu et al., 2010).
Les SNPs présentant la différenciation la plus élevée sont localisés sur le facteur de transcription EPAS1 connu pour être activé en condition d’hypoxie. À la suite de ces scans de différenciation, il a été montré à l’aide de la statistique \(D\) (Table 1.3) que les variants adaptatifs sur EPAS1 ont été transmis grâce à l’introgression issue de l’Homme de Denisova (H. Hu et al., 2017).
| Chr | début | fin | \(D^{*}\) | Gènes |
|---|---|---|---|---|
| 7 | 26800001 | 27000000 | 6.25 | SKAP2 |
| 2 | 46400001 | 46600000 | 6.15 | PRKCE, EPAS1 |
| 2 | 47600001 | 47800000 | 5.92 | MIR559, MSH2, EPCAM, KCNK12 |
| 5 | 200001 | 400000 | 4.99 | CCDC127, PDCD6, SDHA |
| 12 | 8800001 | 9000000 | 4.63 | MFAP5, RIMKLB, A2ML1 |
| 4 | 100400001 | 100600000 | 4.58 | TRMT10A, C4orf17, MTTP |
Principalement réalisés sur des données humaines, les scans génomiques se sont progressivement étendus à d’autres espèces, modèles et non-modèles (Haasl & Payseur, 2016), et les SNPs sont désormais les marqueurs les plus utilisés (Figure 1.9).
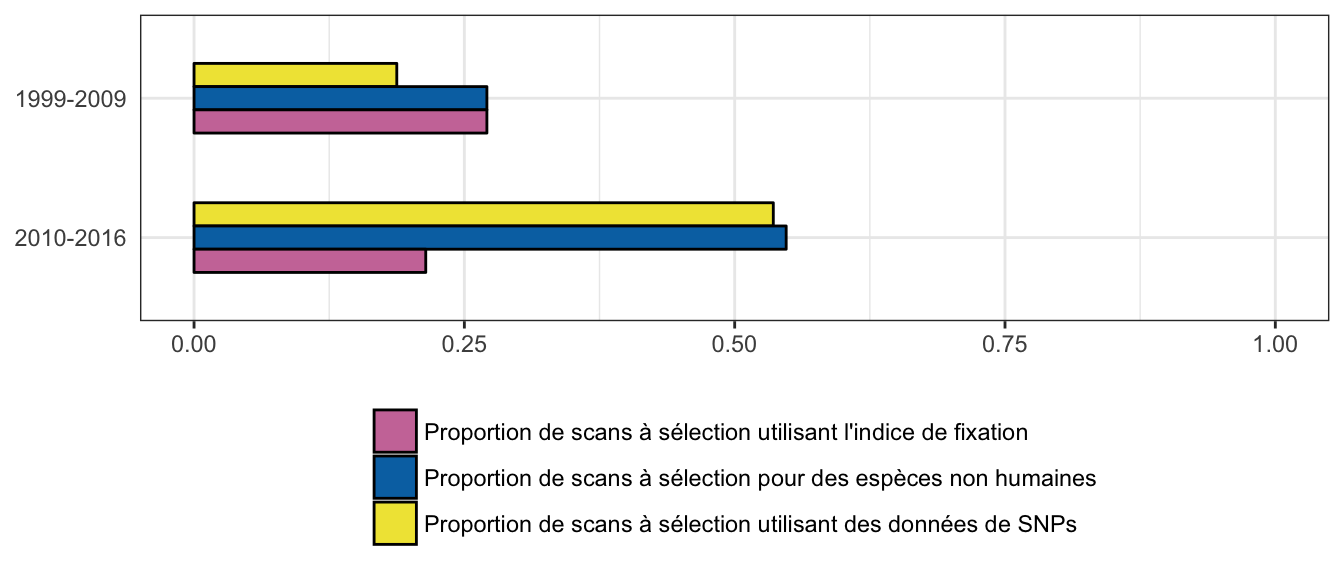
Figure 1.9: Évolution de la proportion d’articles scientifiques s’intéressant à des espèces non humaines et de la proportion de ceux dont l’étude est réalisée sur des données de SNPs, dans le cadre des scans génomiques pour la sélection (Haasl & Payseur, 2016).
Toutefois, certains outils statistiques ayant été développés ne sont plus adaptés. La raison principale étant que les temps de calculs requis par ces outils pour l’analyse de données massives sont souvent prohibitifs. Les méthodes d’analyse exploratoire nécessitent souvent d’être expérimentées plusieurs fois. D’une part parce que le prétraitement des données peut être est amené à changer au cours de l’étude (filtration de marqueurs génétiques présentant une proportion trop élevée de données manquantes, retrait d’individus marginaux, etc.). D’autre part car les méthodes d’analyse proposent généralement un éventail de paramètres dont le choix ne s’impose pas spontanément. Expérimenter une même méthode avec différents critères de qualité et différents paramètres est souvent nécessaire pour minimiser les biais d’utilisation. Tout ceci justifie le recours à des méthodes rapides et adaptées à ces nouveaux volumes de données. Pour satisfaire à ces critères, des méthodes basées sur l’Analyse en Composantes Principales ont été proposées pour étudier l’adaptation locale (Duforet-Frebourg, Luu, Laval, Bazin, & Blum, 2015; Galinsky et al., 2016).
1.4.1 Résultats principaux et organisation du manuscript
Sur la base du travail de thèse de Nicolas Duforet-Frebourg (Duforet-Frebourg, 2014), nous avons cherché dans un premier temps à améliorer la méthodologie statistique implémentée dans le logiciel PCAdapt, ainsi que les performances computationnelles, afin de garantir la possibilité de l’utiliser sur des génômes plus denses et disposant d’une plus grande résolution. Afin de permettre la publication de l’article (Duforet-Frebourg et al., 2015), j’ai réalisé des simulations de modèles en îles et de divergence afin de valider l’approche basée sur la communalité pour réaliser des scans à sélection. J’ai ensuite développé la librairie R pcadapt et proposé une statistique de scan à sélection plus puissante que la communalité qui est la distance robuste de Mahalanobis. Ce travail correspond à la publication (Luu, Bazin, & Blum, 2017). La dernière contribution principale de ma thèse correspond au développement d’une méthode statistique basée sur l’ACP qui permet d’identifier les régions du génôme impliquées dans les évènements d’introgression adaptative. Ce travail correspond au manuscript Scanning genomes for adaptive introgression using principal component analysis et fera l’objet d’une soumission prochainement.
Dans le chapitre 2, nous commençons par décrire les différents modèles, historiques et contemporains, utilisés pour réaliser des scans génomiques pour la sélection. Ce premier chapitre rappelle ensuite quelle est l’utilisation de l’Analyse en Composantes Principales en génétique des populations. La fin du chapitre 2 rappelle les résultats obtenus avec la communalité et la distance de Mahalanobis, qui sont implémentées dans la librairie pcadapt, et qui correspondent aux publications (Duforet-Frebourg et al., 2015; Luu et al., 2017). Le chapitre 3 traite de l’introgression adaptative. Les chapitres 2 et 3 montrent comment l’ACP peut être exploitée de façon à détecter des signaux de sélection ainsi que des régions d’introgression adaptative. Enfin, nous terminerons avec les aspects computationnels et numériques qui nous ont préoccupé tout au long de la thèse, en présentant quelques comparatifs de performances réalisés avec divers algorithmes.
References
Cavalli-Sforza, L. (1994). Francesco. qui sommes-nous? Une histoire de diversité humaine. Trans. Brun, Françoise. Flammarion Ed. Paris: Centre National Des Lettres.
Gillespie, J. H. (2010). Population genetics: A concise guide. JHU Press.
Roll-Hansen, N. (2014). The holist tradition in twentieth century genetics. wilhelm johannsen’s genotype concept. The Journal of Physiology, 592(11), 2431–2438.
Darwin, C. (1980). L’Origine des espèces, trad. Edmond Barbier (1876), Paris, Maspero.
Gayon, J. (1992). Darwin et l’après-darwin: Une histoire de l’hypothèse de sélection dans la théorie de l’évolution. Kimé.
Bateson, W., & Mendel, G. (1913). Mendel’s principles of heredity. University press.
Kimura, M. (1983). The neutral theory of molecular evolution. Cambridge University Press.
Bromham, L., & Penny, D. (2003). The modern molecular clock. Nature Reviews. Genetics, 4(3), 216.
Charlesworth, B., & Charlesworth, D. (2009). Darwin and genetics. Genetics, 183(3), 757–766.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by dna polymorphism. Genetics, 123(3), 585–595.
Drake, J. W., Charlesworth, B., Charlesworth, D., & Crow, J. F. (1998). Rates of spontaneous mutation. Genetics, 148(4), 1667–1686.
Beall, C. M. (2007). Two routes to functional adaptation: Tibetan and andean high-altitude natives. Proceedings of the National Academy of Sciences, 104(suppl 1), 8655–8660.
Jeong, C., & Di Rienzo, A. (2014). Adaptations to local environments in modern human populations. Current Opinion in Genetics & Development, 29, 1–8.
Itan, Y., Powell, A., Beaumont, M. A., Burger, J., & Thomas, M. G. (2009). The origins of lactase persistence in europe. PLoS Computational Biology, 5(8), e1000491.
Harrison, R. G., & others. (1990). Hybrid zones: Windows on evolutionary process. Oxford Surveys in Evolutionary Biology, 7, 69–128.
Stevison, L. (2008). Hybridization and gene flow. Nature Education, 1(1), 111.
Song, Y., Endepols, S., Klemann, N., Richter, D., Matuschka, F.-R., Shih, C.-H., … Kohn, M. H. (2011). Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Current Biology, 21(15), 1296–1301.
Hufford, M. B., Lubinksy, P., Pyhäjärvi, T., Devengenzo, M. T., Ellstrand, N. C., & Ross-Ibarra, J. (2013). The genomic signature of crop-wild introgression in maize. PLoS Genetics, 9(5), e1003477.
Kawecki, T. J., & Ebert, D. (2004). Conceptual issues in local adaptation. Ecology Letters, 7(12), 1225–1241.
Wetterstrand, K. A. (2013). DNA sequencing costs: Data from the nhgri genome sequencing program (gsp).
Muir, P., Li, S., Lou, S., Wang, D., Spakowicz, D. J., Salichos, L., … others. (2016). The real cost of sequencing: Scaling computation to keep pace with data generation. Genome Biology, 17(1), 53.
Gogol-Döring, A., & Chen, W. (2012). An overview of the analysis of next generation sequencing data. Next Generation Microarray Bioinformatics: Methods and Protocols, 249–257.
Xu, S., Li, S., Yang, Y., Tan, J., Lou, H., Jin, W., … others. (2010). A genome-wide search for signals of high-altitude adaptation in tibetans. Molecular Biology and Evolution, 28(2), 1003–1011.
Hu, H., Petousi, N., Glusman, G., Yu, Y., Bohlender, R., Tashi, T., … others. (2017). Evolutionary history of tibetans inferred from whole-genome sequencing. PLoS Genetics, 13(4), e1006675.
Haasl, R. J., & Payseur, B. A. (2016). DETECTING selection in natural populations: MAKING sense of genome scans and towards alternative solutions: Fifteen years of genomewide scans for selection: Trends, lessons and unaddressed genetic sources of complication. Molecular Ecology, 25(1), 5.
Duforet-Frebourg, N., Luu, K., Laval, G., Bazin, E., & Blum, M. G. (2015). Detecting genomic signatures of natural selection with principal component analysis: Application to the 1000 genomes data. Molecular Biology and Evolution, 33(4), 1082–1093.
Galinsky, K. J., Bhatia, G., Loh, P.-R., Georgiev, S., Mukherjee, S., Patterson, N. J., & Price, A. L. (2016). Fast principal-component analysis reveals convergent evolution of adh1b in europe and east asia. The American Journal of Human Genetics, 98(3), 456–472.
Duforet-Frebourg, N. (2014). Statistiques bayésiennes en génétique des populations: Modèle à facteurs et processus gaussiens pour étudier la variation génétique neutre et adaptative (PhD thesis). Grenoble.
Luu, K., Bazin, E., & Blum, M. G. (2017). Pcadapt: An r package to perform genome scans for selection based on principal component analysis. Molecular Ecology Resources, 17(1), 67–77.
Le pastoralisme décrit la relation interdépendante entre les éleveurs, leurs troupeaux et les milieux exploités.↩
épisode de divergence d’une espèce ayant conduit à la formation d’au moins une nouvelle espèce.↩
présentant des variations à l’échelle de l’espèce.↩
SNP (Single-Nucleotide Polymorphism) : polymorphisme d’un seul nucléotide.↩
se présente généralement sous la forme d’un graphique, appelé Manhattan plot, représentant la statistique de test utilisée en fonction de l’emplacement chromosomique.↩